CDCgov / Sars Cov 2_sequencing
Programming Languages
SARS-CoV-2 Sequencing Resources
This document repository is meant to serve as the start of a crowd-sourced collection of information, documentation, protocols and other resources for public health laboratories intending to sequence SARS-CoV-2 coronavirus samples in the coming weeks. This is admittedly a limited first draft, but will continued to collate useful information as additional protocols, tools, and resources are added, and as best practices are identified. While some of the resources here are directed specifically to US state and local public health laboratories in support of diagnostic testing, sequencing and response, we hope that this is a useful resource for the global laboratory community, as we respond to this pandemic threat.
This collection is maintained and curated by Duncan MacCannell from the Office of Advanced Molecular Detection (AMD) at the Centers for Disease Control and Prevention (CDC). Please feel free to suggest additions, edits, clarifications and corrections -- either by posting an issue, filing a pull request or by contacting me directly by email or twitter. In the meantime, I'll continue to add and mirror useful resources here as they become available.
INDEX
- Sequencing Protocols
- Bioinformatic Tools, Scripts and Workflows
- Quality Management
- Submitting to Public Sequence Repositories
- Linking Sequence Accessions
- Other Useful References and Resources
- Notices and Disclaimers
Disclaimer
The findings and conclusions in this document and the attendant repository are those of the author and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Use of trade names is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention or by the U.S. Department of Health and Human Services.
Sequencing Protocols
1. OXFORD NANOPORE
The following sequencing protocols, checklists and job-aids are primarily designed for the Oxford Nanopore MinION, and have been kindly shared by research groups throughout the world (please see individual protocols for attribution and citing purposes). Even so, most of these protocols should scale to larger ONT instruments without significant modifications.
a) ARTIC Network nCoV-2019 Sequencing Protocol
This protocol was developed and released by the fine folks at ARTIC Network, and was subsequently refined based on comments from Itokawa et al, which identified potential issues and proposed an alternate L18 primer.
-
ARTIC on Illumina - Complete Walk-through - Grubaugh/Andersen/Loman labs
-
Integrated bioinformatics (RAMPART) - documentation below under bioinformatics methods.
UPDATE: Recent preprint by Tyson et al. and the ARTIC Network team describes a greatly streamlined workflow for ARTIC-Nanopore sequencing of SARS-CoV-2, including functional multiplexing library construction up to 96 samples. Protocol and details are here.
b) Doherty Institute VIDRL Sequencing Protocols
The Victorian Infectious Diseases Reference Laboratory (VIDRL) at the Peter Doherty Institute for Infection and Immunity released two protocols for the ONT MinION, which they successfully used to sequence early Australian SARS-CoV-2 samples.
2. ILLUMINA
a) Illumina Nextera Flex Enrichment Sequencing Protocol
Illumina's Research and Development group has recently developed and validated a custom, research use only (RUO) enrichment sequencing strategy based on their Nextera Flex chemistry.
b) SARS-CoV-2 Enrichment Sequencing by Spiked Primer MSSPE (NIAID/UCSF/CZBioHub)
The NIAID laboratory team in Cambodia, in collaboration with UCSF, CZBioHub and IPC, has released a metagenomic sequencing with spiked primer enrichment (MSSPE) protocol for SARS-CoV-2. The protocol is available on protocols.io.
c) Illumina Shotgun Metagenomics Sequencing Protocol
Illumina's technical note on sequencing coronavirus samples using a comprehensive metagenomic sequencing approach was one of the earlier protocols released for SARS-CoV-2, and remains an effective option for shotgun sequencing.
d) SARS-CoV-2 and related virus sequencing with capture enrichment (Broad Institute)
The Sabeti lab, at the Broad Institute, released a probe set for comprehensive whole-genome capture of SARS-CoV-2 and respiratory-related viruses (human-infecting coronaviruses, HRSV, HMPV, HPIVs, Human mastadenovirus A-G, Enterovirus A-E, Rhinovirus A/B/C, influenza A/B/C). The probe set is available as V-Respiratory on the probe designs page of the CATCH repository. It was initially released in January, 2020 and most recently updated in March, 2020. Probes can be ordered from Twist Bioscience; we have used the protocol for Twist custom panels with slight modifications for low input Nextera XT libraries.
e) ARTIC on Illumina
A number of different laboratories have implemented derivatives of the ARTIC amplicon scheme on Illumina.
- Protocol from the Grubaugh lab at Yale (gdoc)
- Virgina DCLS has a detailed ARTIC-on-Illumina protocol, including worksheets, flowcharts and helpful hints.
- Joel Sevinsky and the intrepid team at StaPH-B have adapted ARTICv3 for Illumina for public health laboratories that are already configured for PulseNet sequencing. - SARS-CoV-2 Sequencing on Illumina MiSeq using ARTIC Protocol: Part 1 - Tiling PCR - SARS-CoV-2 Sequencing on Illumina MiSeq using ARTIC Protocol: Part 2 - Illumina DNA Flex
Modifications, Improvements and Derivatives of ARTIC
- Freed and Silander developed a modified ARTIC protocol with 1200bp inserts(primers)
- Gohl and collagues at UMN describe an elegant tailed amplicon approach that enables ARTIC without the need for ligation or tagmentation steps (biorXiv)(protocols.io)
- Sorensen, Karst and Knutsson from Alborg University have described a tailed long amplicon derivative of ARTIC (protocols.io)
3. PACIFIC BIOSCIENCES
PacBio maintains a COVID-19 landing page with updated resources on existing protocols and SARS-CoV2-2 assay development recommendations.
Barcoding options
a) Sinai 1.5kb and 2kb tiled amplicon protocol and PacBio barcoding options b) Eden 2.5kb tiled amplicon protocol and PacBio barcoding options c) CDC 500bp and 900bp tiled amplicon protocol and PacBio barcoding options
Files are mirrored here.
4. IONTORRENT
A number of laboratories have reported success with the Ion AmpliSeq SARS-CoV-2 Research panel for the IonTorrent S5 platform. Amplicon strategies, such as ARTIC, should also work for the S5, and we'd welcome the addition of any working protocols and other resources to this section.
- Ion AmpliSeq SARS-CoV-2 Research Panel for GeneStudio S5
- Ion AmpliSeq SARS-CoV-2 Research Panel for the Genexus System
5. TANDEM
a) SARS-CoV-2 Parallel Sequencing by Illumina and ONT (UWMadison ZEST)
Staff and students from Thomas Friedrich and Dave O'Connor's laboratories at UWMadison have put together a tandem sequencing protocol and bioinformatic workflow that incorporates Illumina and ONT sequence. While this may be overkill for routine or high-throughput public health purposes, the necessary protocols, scripts and documentation are available here.
Bioinformatic Tools, Scripts and Workflows
0. General Resources.
A few different sites have resources that are foundational to most bioinformatics analysis.
- NCBI and EBI are both INSDC public repositories, and contain all public access sequences, annotation, analysis and derived data. NCBI has added a specific landing page for SARS-CoV-2 research resources, available here.
- GISAID was put into place to provide a framework for open sharing of influenza sequence data, while maintaining strict governance over the use of data and attribution to sequence submitters.
- The original SARS-CoV-2 reference sequence, "Wuhan-Hu-1", submitted by Zhang Yongzhen and colleagues is mirrored from NCBI in the
sequencesfolder here. In that folder, you will also find a dated set of public genomes in that folder, downloaded from NCBI on a semi-regular basis. NB: These are consensus sequences manually pulled from NCBI Genbank. More complete sets of sequences are available at the GISAID public access repository, which requires account registration and adherence to a strict code of conduct. - Genexa has put together a page with precomputed kmer sets, indicies, reference sequences, and a number of other useful resources for bioinformatic analysis of SARS-CoV-2 NGS data.
- Illumina has just released a SARS-CoV-2 Software Toolkit, including premade RT-PCR and sequencing workflows for ClarityLIMS, FPGA (DRAGEN)-powered bioinformatics, and direct submission to GISAID.
1. CDC NCIRD/DVD Bioinformatics SOPs
This section describes the basic bioinformatic workflow that the Viral Discovery laboratory in NCIRD, and other teams at CDC use for quality assessment, assembly and comparison of coronavirus sequences. IRMA, the Iterative Refinement Meta-Assembler developed by CDC's Influenza Division for routine influenza surveillance, has recently been updated to support both ebolavirus and coronavirus assembly tasks. While IRMA isn't used for all SARS-CoV-2 assemblies at CDC, it is a powerful tool for complex or problematic samples and datasets.
- Preprint describing
- IRMA: Iterative Refinement Meta-Assembler is available here.
- CLCbio (Jonathan Jacobs) has released workflows for the NCIRD/DVD protocols on Illumina and Nanopore. Older versions available here.
2. CLCbio Genomics Workbench
QIAGEN has released example workflows and tutorials for analyzing Illumina and Oxford Nanopore SARS-CoV-2 sequence data using CLC Genomics Workbench v20.0.3. Note - these workflows are "Research Use Only" (RUO), and may need to modified to fit upstream protocols. Free temporary licenses for CLC GWB and IPA are available, as well as a series of webinars and tutorials are available to familiarize users with the workflows. Jonathan Jacobs and Leif Schauser are available for user support and specific questions.
- Temporary licenses for CLC Genomics Workbench and Ingenuity Pathway Analysis
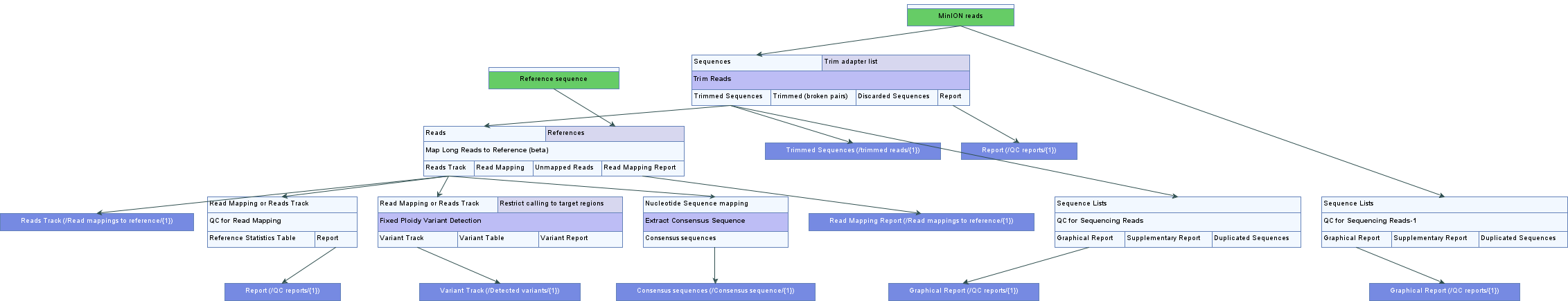
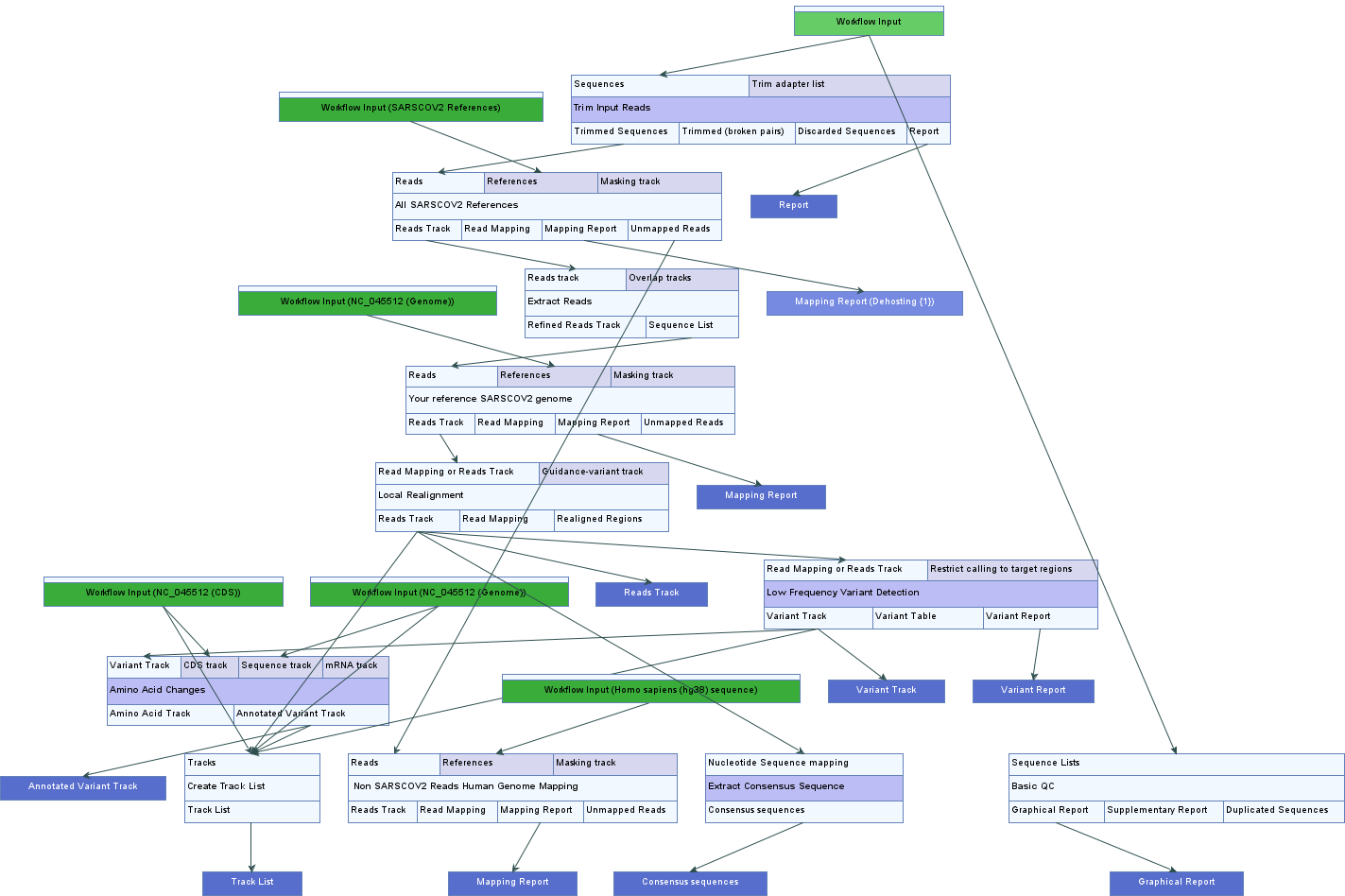
- SARS-CoV-2 Analysis Tutorial with Nanopore Data (Figure)
- SARS-CoV-2 Workflow and Tutorial Data for Nanopore Data (Figure)
- CLC Illumina Workflow v1
- Additional CLC Genomics Workbench Workflows developed by Jonathan Jacobs for QIAseq SARS-CoV-2 panel, Ion AmpliSeq SARS-COV-2 panel, and ARTICv3 Nanopore & llumina panels.
{kind=link}
{kind=link}
3. ARTIC Network Bioinformatics
The ARTIC Network has released detailed instructions on how to setup and configure the conda environment needed to run their analysis pipelines. These are complete bioinformatic workflows, including runtime visualization, basecalling, mapping/assembly and reporting in a single, portable environment. The artic-nCoV2019 repo includes source code and build instructions for a custom RAMPART configuration. Additional instructions and documentation are available below.
4. BugSeq
BugSeq has added support for automatic SARS-CoV-2 analysis (QC, consensus, variant calling and lineage detection) from nanopore sequencing data. This analysis (example) is triggered by detection of SARS-CoV-2 in data submissions and is automatically tailored for your experimental design (metagenomic and ARTIC v1,v2 and v3 amplicons supported). BugSeq is making analysis of SARS-CoV-2 samples available free of charge for any user publishing their data in a pre-print or the peer-reviewed literature. Additional information and documentation are available below:
5. One Codex
One Codex has added support to its analysis platform for analyzing SARS-CoV-2 samples. This analysis (example) will be automatically run on any samples with SARS-CoV-2 reads. One Codex is making analysis of SARS-CoV-2 samples available free of charge for all users sharing their results and data publicly. Additional information and documentation are available below:
6. Broad viral-ngs tools
The Broad Institute's viral genomics analysis tools can assist with assembly, metagenomics, QC, and NCBI submission prep, for Illumina-generated data on viral genomes. It is available in the following forms:
- The Terra cloud platform (workspace including example SARS-CoV-2 data from SRA, blog post, getting started)
- The DNAnexus cloud platform (workflows)
- The Dockstore tool repository service - integrates with several cloud platforms, or download to run on-prem (workflows)
- Github (workflows)
The tools include:
- denovo and reference based assembly
- short read alignment and coverage plots
- krakenuniq metagenomic classification
- NCBI: SRA download, Genbank annotation download, Genbank submission prep
- multiple alignment of genomes w/MAFFT
- Illumina basecalling & demux, metrics, fastQC, ERCC spike-in counter
7. Genome Detective Virus tool
Genome Detective virus tool does QC, assembly and identification of SARS-CoV-2 from a wide range of sequencing protocols (metagenomic or targeted sequencing).
Raw sequence read files (FASTQ) can be uploaded directly in this web-based tool, and consensus sequences can be subsequently analyzed by the the Coronavirus Typing Tool.
Example output:
8. CosmosID
CosmosID has recently posted a blog entry on their site, describing how to use their web-based analysis platform to analyze SARS-CoV-2 data.
9. ARTIC on Illumina Bioinformatic Workflow
@ErinYoung and Kelly Oakeson at the Utah Department of Health have outlined their bioinformatics approach for SARS-CoV-2 sequences using ARTIC primers, sequenced on Illumina.
10. Galaxy
Workflows are available for the Galaxy Platform
- Cheoinformatics: Virtual screening of the SARS-CoV-2 main protease
- Genomics: Analysis of COVID-19 data using Galaxy, BioConda and public research infrastructure
- ARTIC: Amplicon analysis using Artic workflows
These repositories provide best practise workflows for genomic and chemoinformatic analyses for SARS-CoV-2 data. In addition to providing tools and workflows we provide free public computational infrastrcture for immediate use by anyone worldwide using a consortium of Galaxy instances from US, EU, and Australia.
The chemoinformatics workflows can be used to conduct fragment screening using molecular docking. This has already been done for the SARS-COV-2 main protease (MPro) by the Diamond Light Source's XChem team, InformaticsMatters and the European Galaxy Team. The genomics workflows use entirely open source software and open access platforms to perform (1) data pre-processing, (2) genome assembly, (3) estimation of MRCA timing, (4) analysis of intrahost variation, (5) analysis of substitutions within the S gene, and (6) analysis of recombination and selection.
11. Nanopore Direct RNA Analysis Using MasterOfPores
The Epitranscriptomics and RNA Dynamics Lab (Novoa) and the Bioinformatics Core Facility (BioCore at the CRG have released a set of tools and resources to support the analysis of nanopore direct RNA sequencing data.
12. BioNumerics SARS-CoV-2 Plugin
Applied Maths/bioMerieux have released a plugin for BioNumerics that facilitates the processing and analysis of SARS-CoV-2 genomic sequences, whether downloaded from a public data repository or generated locally. More info on the tool and a tutorial can be found here.
13. StaPH-B ToolKit Monroe Workflow
The State Public Health Bioinformatics (StaPH-B) consortium has made their Monroe workflow accessible through the StaPH-B ToolKit. Monroe consists of three separate Nextflow pipelines for
- ARTIC + Illumina paired-end read assembly
- ARTIC + Oxford Nanopore Technlogies read assembly
- Cluster analysis from assembled SC2 genomes
14. fastv: identify SARS-CoV-2 from sequencing data in one minute
Fastv is a little-weight independent tool for ultra-fast identification of SARS-CoV-2 and other microbes from sequencing data. It detects SARS-CoV-2 sequences from FASTQ data, generates JSON reports and visualizes the result in HTML reports. It supports both short reads (Illumina, BGI, etc.) and long reads (ONT, PacBio, etc.). More information can be found here.
15. EDGE COVID-19: A web platform for generating SARS-CoV-2 genomes
EDGE COVID-19 is a standardized web-based workflow for automated reference-based genome assembly of SARS-CoV-2 samples. The workflow accommodates Illumina or Oxford Nanopore Technologies data, performs read mapping and provides static and interactive figures/graphs to explore quality and any discovered SNPs, Variants, Gaps, and indels. Given raw FASTQ file(s) from amplicon-based methods (ARTIC, CDC) or shotgun sequencing (including from enrichment protocols), EDGE COVID-19 automates the production of a SARS-CoV-2 genome that is ready for submission to GISAID or GenBank. We have automated the process to submit high quality genomes to GISAID (with required metadata) and we are working on a similar process for SRA and GenBank.
Here is a link to the arXiv paper describing the workflow.
Our other COVID-19 related efforts can be found here.
16. MiCall: Pipeline for processing NGS data to genotype human RNA viruses like SARS-CoV-2, HIV and hepatitis C
MiCall processes NGS read data from platforms like Illumina by either assembling them or mapping them to a set of reference sequences. Then, it reports consensus sequences, variant mixtures, and quality control reports. For HIV and hepatitis C, it also reports drug resistance interpretations of the variant mixtures.
MiCall is open-source software and comes packaged to be run under Docker or Singularity, to make installation easy. It's also available to run on Illumina's BaseSpace web service, but the SARS-CoV-2 support is not yet available there.
17. EzCOVID19: A bioinformatics platform for rapid detection, identification, and characterization of the SARS-CoV-2 virus
ChunLab/EzBiome has developed a cloud-based bioinformatics platform, EzCOVID19, for rapid detection, identification, and characterization of the SARS-CoV-2 virus from raw metagenomic, metatranscriptomic, RNA-seq, and/or isolate (amplicon or enrichment) NGS data suspected of containing the SARS-CoV-2 virus. EzCOVID19 provides scientists with a consensus genome assembly along with statistics related to genome coverage, depth metrics, and coverage plots relative to the reference SARS-CoV- 2 genome. EzCOVID19 enables characterization and typing of the entire viral genome, when the user obtains adequate coverage of the genome. It provides Single Nucleotide Variant (SNV) information, including a graph and table with detected variants in the SARS-CoV-2 genome, identifies most similar genomes available in the reference databases, based on alignment statistics and SNVs, including a maximum likelihood or parsimony based similarity tree decorated with SNV profiles. It also offers classification or typing of the queried genome using EzBioCloud’s SNP based classification scheme of SARS-CoV-2 variants, including an evolutionary analysis of the detected SARS-CoV-2 type along with other types observed among publicly available SARS-CoV-2 genomes.
Here is an example of EzCOVID19 analysis outputs. Please contact [email protected] for any questions or queries.
18. Chan Zuckerberg BioHub/CZI IDSeq
The Chan Zuckerberg BioHub and Chan Zuckerberg Initiative have put together updates to the IDSeq platform, enabling users to analyze MSSPE or ARTICv3 amplicon SARS-CoV-2 sequence data, in addition to their existing tools for metagenomic analysis. They have also provided a number of workflows over on their Github site, including an automated pipeline for building SARS-CoV-2 consensus genomes WDL link.
Quality Management
This section will describe best practices for laboratory and bioinformatic quality assurance, including preflight checks for sequence and metadata submission to public repositories.
1. FDA ARGOS SARS-CoV-2 Reference Sequence Data and Materials
The FDA, in collaboration with CDC, BEIR, UMaryland IGS, and others, have recently put together reference sequences, and materials for NGS sequencing and assay development. These resources will be invaluable to many laboratories implementing NGS quality management programs.
2. Host Sequence Removal
SanitizeMe is a set of scripts and an X11 GUI for removing human host sequences from metagenomic data before SRA submission. Software and documentation are available here.
- to do: I'd love for people to help describe their actual QC processes.
3. In silico SARS-Cov-2 benchmark dataset
This benmark dataset is generated from Lemieux et al, 2020. The tree should resolve to three clades and an outgroup X11.
Submitting to Public Sequence Repositories
WHO Code of Conduct: Sequence Sharing During Outbreaks
The draft WHO code of conduct for open and timely sharing of pathogen genetic sequence data during outbreaks of infectious disease lays out an important and sensible set of principles for sharing pathogen genetic sequence data during outbreaks of international importance. The text is available here.
Sequence naming conventions for public repositories
We are proposing simplified naming conventions for sequences submitted to GISAID and NCBI from US public health and clinical laboratories.
| COUNTRY | / STATE-LAB-SAMPLE / | YEAR |
|---|---|---|
USA |
/ CA-CDPH-S001 / |
2020 |
USA |
/ UT-UPHL-0601 / |
2020 |
USA |
/ AZ-TGEN-N1045 / |
2020 |
USA |
/ ID-UW-0316 / |
2020 |
The proposed convention is as follows: 1) country (USA). 2) The middle sample identification cell should include two-letter state (eg: CA), an abbreviated identifier for the submitting lab (eg: CDPH), as desired, and a unique sequence identifier (eg:01, S01, 454, ...), with all three terms separated by hyphens.
For states with only one submitting laboratory (which should be most), the identifier for the submitting laboratory may be omitted, resulting in a simple, state-level identifier such as USA/UT-573/2020.
These recommendations are roughly compatible with existing submissions to GISAID and NCBI, but are completely open for debate. The current ICTV recommendations are here, with the original biorXiv here.
Recommended formatting and criteria for sample metadata
NCBI SARS-CoV-2 Genbank/SRA
The National Center for Biotechnology has established a custom landing page for SARS-CoV-2 sequences and data, and is working to develop streamlined submission processes for Genbank and SRA. For the time being, we suggest basing metadata and submission formatting on GISAID EpiCoV, which tends to be more comprehensive and structured. We will develop specific guidance for NCBI submissions. In the meantime, here are some general resources to help with NCBI data submission and metadata management.
1. NCBI Submission Portal
Individual sequences can be submitted to NCBI using the following web form. Create an NCBI user account, and select "SARS-CoV-2 (through BankIt)".
NCBI has provided provisional guidance for SARS-CoV-2 sequence submissions to SRA and Genbank. Detailed instructions are available here. Any questions can be directed to NCBI staff here.
2. NCBI Batch Submissions
NCBI has indicated that they plan to develop a specific rapid submission process for SARS-CoV-2 sequences. In the meantime, I believe you should be able to follow the FDA/CFSAN submission protocol below, which includes links to appropriate interfaces and templates (with obvious changes for pathogen and project information).
3. FDA/CFSAN NCBI Submission and Data Curation Protocols
The FDA Center for Food Safety and Applied Nutrition (CFSAN) has released a number of protocols as part of their GenomeTrakr Network that may be useful for NCBI sequence submission and metadata curation. While they are written specifically for laboratories that are conducting routine sequencing of foodborne bacterial pathogens, these protocols provide an overview of sequence submission to the NCBI pathogen portal, metadata and preflight data checks.
- NCBI submission protocol for microbial pathogen surveillance
- Populating the NCBI pathogen metadata template
- NCBI Data Curation - Pending release
4. NIAID METAGENOTE (SRA Submissions)
Many users are already familiar with NIAID's wonderful METAGENOTE submission tool, which is frequently used to get microbial genomic and metagenomic datasets, and corresponding metadata organized and submitted to SRA. The NIAID Bioinformatics team has recently released a walkthrough on how to use the METAGENOTE platform for SARS-CoV-2 sequence read submissions. Detailed instructions here with an instructional video as well.
GISAID EpiCoV
The GISAID EpiCoV Public Access repository is based on existing submission processes and data structures for large-scale influenza surveillance (GISAID EpiFlu). As such, submitters to EpiCoV will discover that several of the required metadata submission fields may be problematic. Nonetheless, a number of laboratories have been submitting sequences with the following:
| METADATA FIELDS (GISAID) | GUIDANCE |
|---|---|
Virus name |
USA/FL-UF-103/2020 (see above) |
Accession ID |
|
Type |
betacoronavirus |
Collection date |
YYYY-MM-DD |
Location |
USA / State / County? |
Additional location information |
|
Host |
Human |
Additional host information |
|
Gender |
(no guidance) |
Patient age |
(no guidance, could be binned) |
Patient status |
(no guidance) |
Specimen source |
(free text) |
Outbreak detail |
omit |
Last vaccinated |
omit |
Treatment |
omit |
At a minimum, we suggest that samples be submitted with collection date location host information attached. location, host, gender patient age are all required fields, and several of them likely constitute personally-identifiable information. While they cannot be left blank for submission, you can submit the record successfully (in both single or batch mode) by entering "unknown".
Note that for GISAID submissions, users must register for an account, and must successfully submit a single submission before being granted access to the bulk submission template and interface.
A copy of the current bulk submission template is available here.
Linking Sequence Accessions
For data linkage, we are proposing the following template, as a simple, lightweight line list of tab-separated values. If this consensus recommendation for data linkage is acceptable, a preformatted .TSV will be made available. We recognize that not all samples sent for sequencing have a PUID associated.
| SEQUENCE_NAME | GISAID_ID | GENBANK_ID | COLLECTION_DATE | PUID/COVID-ID |
|---|---|---|---|---|
| USA/CA-CDPH-999/2020 | EPI_ISL_999999 | MT99999999 | 2020-04-01 | 99999 |
In this simple proposed schema, GISAID ID or GENBANK ID and COLLECTION DATE are required fields, and our hope is to maximize PUID completion. All accession numbers, including PUID should be entered without any superfluous text or annotation.
Other Useful References and Resources
SARS-CoV-2 Genomics Consortia
Useful References
- Open-source analytics tools for studying the COVID-19 coronavirus outbreak
- NU COVID-19 Resource Collection
- Global Initiative on Open-source Genomics for SARS-CoV-2
COVID-19 Host Genomics
Slides and Presentations
Visualization and Phylogenetics
- Nextstrain hCoV-19
- UCSC SARS-CoV-2 browser
- CNCB 2019 Novel Coronavirus Resource (2019nCoVR)
- PANGO lineages
- Plylodynamics of SARS-CoV-2-GISAID subsamples
- US SPHERES Nextstrain builds
CDC COVID-19 Genomic Epidemiology Toolkit
Diagnostic Resources
- SARS-CoV-2 PCR Primer List (Grubaugh Lab, Yale SPH)
- EDGE Bioinformatics: In-Silico Evaluation of Diagnostic Assays (LANL)
Additional Protocol Resources
Notices and Disclaimers
Public Domain
This repository constitutes a work of the United States Government and is not subject to domestic copyright protection under 17 USC § 105. This repository is in the public domain within the United States, and copyright and related rights in the work worldwide are waived through the CC0 1.0 Universal public domain dedication. All contributions to this repository will be released under the CC0 dedication. By submitting a pull request you are agreeing to comply with this waiver of copyright interest.
License
Unless otherwise specified, the repository utilizes code licensed under the terms of the Apache Software License and therefore is licensed under ASL v2 or later.
This source code in this repository is free: you can redistribute it and/or modify it under the terms of the Apache Software License version 2, or (at your option) any later version.
This source code in this repository is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the Apache Software License for more details.
You should have received a copy of the Apache Software License along with this program. If not, see http://www.apache.org/licenses/LICENSE-2.0.html
Any source code forked from other open source projects will inherit its license.
Privacy
This repository contains only non-sensitive, publicly available data and information. All material and community participation is covered by the Disclaimer and Code of Conduct. For more information about CDC's privacy policy, please visit http://www.cdc.gov/other/privacy.html.
Contributing
Anyone is encouraged to contribute to the repository by forking and submitting a pull request. (If you are new to GitHub, you might start with a basic tutorial.) By contributing to this project, you grant a world-wide, royalty-free, perpetual, irrevocable, non-exclusive, transferable license to all users under the terms of the Apache Software License v2 or later.
All comments, messages, pull requests, and other submissions received through CDC including this GitHub page may be subject to applicable federal law, including but not limited to the Federal Records Act, and may be archived. Learn more at http://www.cdc.gov/other/privacy.html.
Records
This repository is not a source of government records, but is a copy to increase collaboration and collaborative potential. All government records will be published through the CDC web site.
Updated: 20201013 @dmaccannell