theislab / Scgen
Programming Languages
Projects that are alternatives of or similar to Scgen
scGen 



Introduction
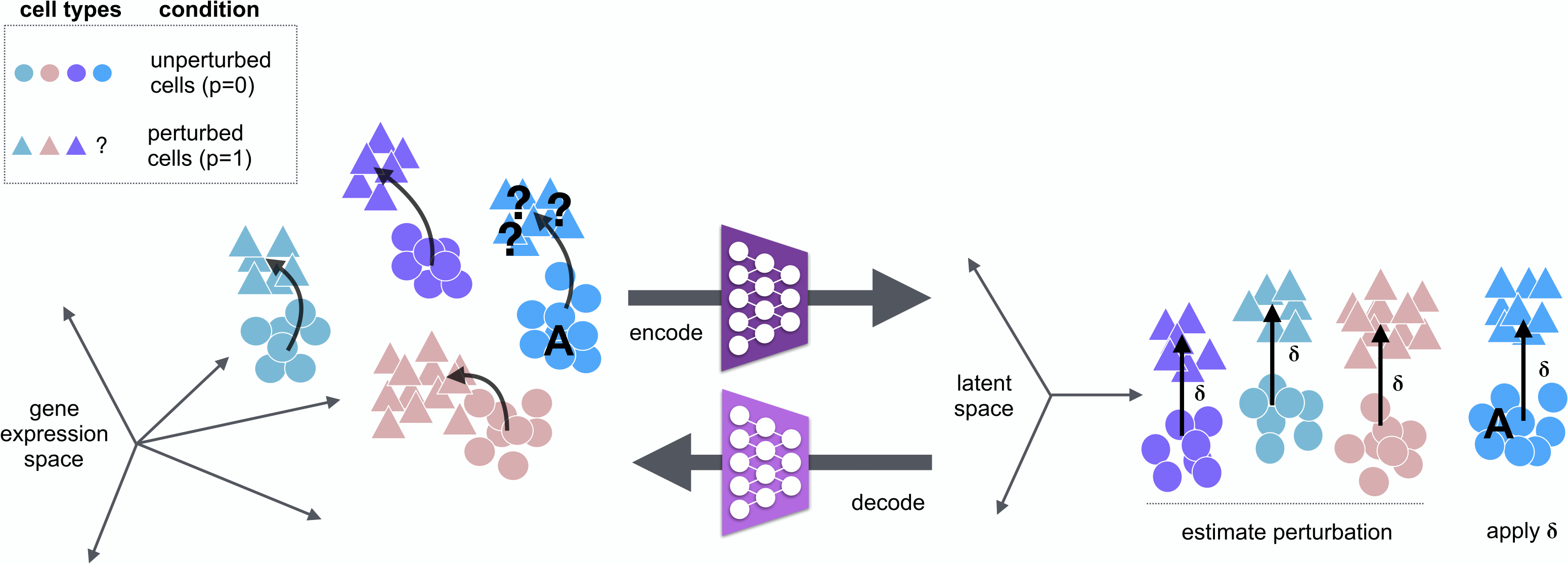
A tensorflow implementation of scGen. scGen is a generative model to predict single-cell perturbation response across cell types, studies and species (Nature Methods, 2019) .
Getting Started
*What you can do with scGen:
-
Train on a dataset wih multiple cell types and conditions and predict the the perturbation effect on the cell type which you only have in one condition. This scenario can be extended to multiple species where you want to predict the effect of a specific species using another or all the species.
-
Train on a dataset where you have two conditions (e.g. control and perturbed) and predict on second dataset with similar genes.
-
Remove batch effect on labeled data. In this scenario you need to provide cell_type and batch labels to the method. Note that
batch_removaldoes not require all cell types to be present in all datasets (batches). If you have dataset specific cell type it will preserved as before. -
We assume there exist two conditions in you dataset (e.g. control and perturbed). You can train the model and with your data and predict the perturbation for the cell type/species of interest.
-
We recommend to use normalized data for the training. A simple example for normalization pipeline using scanpy:
import scanpy as sc
adata = sc.read(data)
sc.pp.normalize_per_cell(adata)
sc.pp.log1p(adata)
- We further recommend to use highly variable genes (HVG). For the most examples in the paper we used top ~7000 HVG. However, this is optional and highly depend on your application and computational power.
Installation
Installation with pip
To install the latest version from PyPI, simply use the following bash script:
pip install scgen
or install the development version via pip:
pip install git+https://github.com/theislab/scgen.git
or you can first install flit and clone this repository:
pip install flit
git clone https://github.com/theislab/scGen
cd scgen
flit install
On Windows machines you may need to download a C++ compiler if you wish to build from source yourself.
Notes for installing:
- If you plan to use GPU, please consider installing tensorflow-GPU after installing scgen.
- We recommend using tensorflow < 2 and compatible Keras versions and python 3.8, you might have to change
a few thing to use tf 2.0, for more info see here.
*If you get error such as
No matching distribution found for tensorflowwhile installing, please check your python version and install right tf and keras version for that (we recommend to use python <=3.7)
Examples
-
For perturbation prediction check this example for interferon (IFN)-β stimulation from Kang et al..
-
For batch-removal check our example on integrating four pancreas datasets.
Reproducing paper results
In order to reproduce paper results visit here.
References
Lotfollahi, Mohammad and Wolf, F. Alexander and Theis, Fabian J. "scGen predicts single-cell perturbation responses." Nature Methods, 2019. pdf