We have a new bioinformatic resource that largely replaces the functionality of this project! See our new repository here: https://github.com/nanoporetech/pipeline-nanopore-ref-isoforms
This repository is now unsupported and we do not recommend its use. Please contact Oxford Nanopore: [email protected] for help with your application if it is not possible to upgrade to our new resources, or we are missing key features.
Pipeline for annotating genomes using long read transcriptomics data with pinfish
Pinfish is a collection of tools helping to make sense of long transcriptomics data (long cDNA reads, direct RNA reads). Pinfish is largely inspired by the Mandalorion pipeline. It is meant to provide a quick way for generating annotations from long reads only and it is not meant to provide the same functionality as pipelines using a broader strategy for annotation (such as LoReAn).
This snakemake pipeline runs the pinfish tools to generate GFF2 annotations from a reference genome and input long reads.
Getting Started
Input
-
The input reads must be in fastq format. The default parameters in
config.ymlare tuned for stranded data. If your input is unstranded cDNA data, it is recommended to run pychopper on the input fastq in order to detect the strandedness of the reads. It is recommended to runpychopperfor stranded cDNA data as well to select for reads which have both the reverse transcription and the strand switching primer. -
The input genome must be in fasta format.
Output
The pipeline produces the following output:
alignments/reads_aln_sorted.bam- the input reads aligned to the input genome byminimap2in BAM format.results/raw_transcripts.gff- the spliced alignments converted into GFF2 format (one transcript per reads)results/clustered_transcripts.gff- The transcripts resulting from the clustering process bycluster_gff.results/clustered_transcripts_collapsed.gff- The transcripts resulting from the clustering with the likely degradation artifacts filtered out.results/polished_transcripts.fas- The sequences of the polished transcripts (one per cluster) produced bypolish_clusters.alignments/polished_reads_aln_sorted.bam- The spliced alignment of the polished transcripts to the input genome.results/polished_transcripts.gff- The alignments of the polished transcripts converted into GFF2 format.results/polished_transcripts_collapsed.gff- The polished transcripts GFF with the likely degradation artifacts filtered out.results/corrected_transcriptome_polished_collapsed.fas- The reference corrected transcriptome generated from the input genome andpolished_transcripts_collapsed.gff.- For all practical purposes
results/polished_transcripts_collapsed.gffis the final output of the pipeline and likely to be the most accurate.
Depedencies
- miniconda
- snakemake - easily installed via conda
- minimap2 - installed by the pipeline via conda
- samtools - installed by the pipeline via conda
- racon - please install from source!
Layout
README.mdSnakefile- master snakefileconfig.yml- YAML configuration filesnakelib/- snakefiles collection included by the master snakefilepinfish/- pinfish source directory
Installation
Clone the pipeline and the pinfish toolset by issuing:
git clone --recursive https://github.com/nanoporetech/pipeline-pinfish-analysis.gitUsage
Edit config.yml to set the input genome, input fastq and parameters, then issue:
snakemake --use-conda -j <num_cores> allResults
Performance on SIRV E0 mix spike-in data
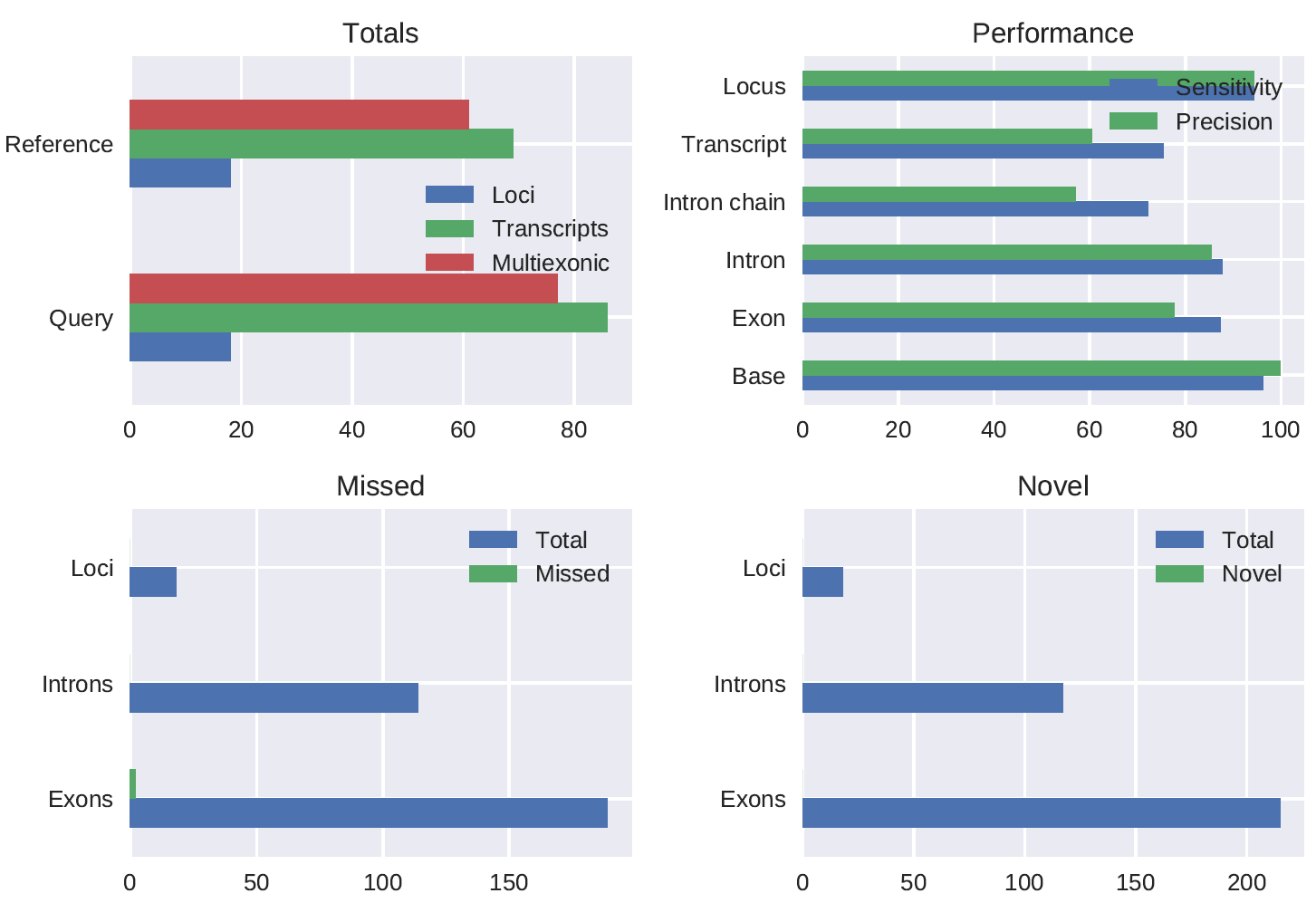
A SIRV E0 mix stranded 1D PCR cDNA (chemistry not yet released) spike-in dataset preprocessed using pychopper produced 786844 full length reads (62.3% of total reads) of which 97% was mapped. The gffcompare comparison of the polished_transcripts_collapsed.gff output of the pipeline run with config_SIRV.yml to the true SIRV annotation gave the following results:
#= Summary for dataset: polished_transcripts_collapsed.gff
# Query mRNAs : 86 in 18 loci (77 multi-exon transcripts)
# (10 multi-transcript loci, ~4.8 transcripts per locus)
# Reference mRNAs : 69 in 18 loci (61 multi-exon)
# Super-loci w/ reference transcripts: 18
#-----------------| Sensitivity | Precision |
Base level: 96.2 | 99.8 |
Exon level: 87.3 | 77.7 |
Intron level: 87.7 | 85.5 |
Intron chain level: 72.1 | 57.1 |
Transcript level: 75.4 | 60.5 |
Locus level: 94.4 | 94.4 |
Matching intron chains: 44
Matching transcripts: 52
Matching loci: 17
Missed exons: 2/189 ( 1.1%)
Novel exons: 0/215 ( 0.0%)
Missed introns: 0/114 ( 0.0%)
Novel introns: 0/117 ( 0.0%)
Missed loci: 0/18 ( 0.0%)
Novel loci: 0/18 ( 0.0%)
Total union super-loci across all input datasets: 18
86 out of 86 consensus transcripts written in gffcmp.annotated.gtf (0 discarded as redundant)
Performance on real data
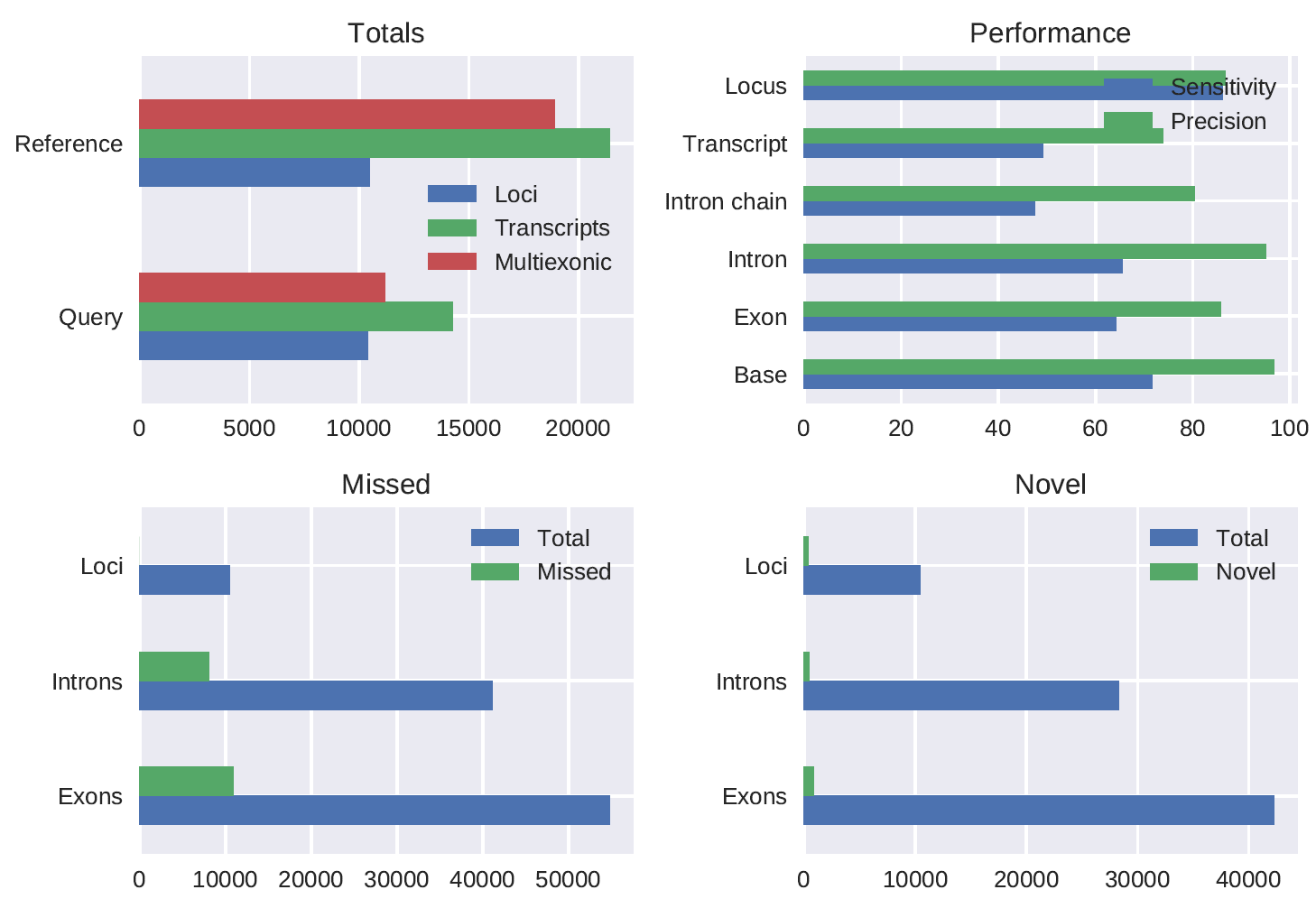
A Drosophila melanogaster stranded 1D PCR cDNA (chemistry not yet released) dataset preprocessed using pychopper produced 7843107 full length reads (52.2% of total reads) of which 95.6% was mapped. The gffcompare comparison (using the -R flag) of the polished_transcripts_collapsed.gff output of the pipeline run with config_Dmel.yml to the Ensembl annotation gave the following results:
#= Summary for dataset: polished_transcripts_collapsed.gff
# Query mRNAs : 14264 in 10407 loci (11181 multi-exon transcripts)
# (2091 multi-transcript loci, ~1.4 transcripts per locus)
# Reference mRNAs : 21439 in 10469 loci (18896 multi-exon)
# Super-loci w/ reference transcripts: 9510
#-----------------| Sensitivity | Precision |
Base level: 71.6 | 96.8 |
Exon level: 64.2 | 85.8 |
Intron level: 65.5 | 95.0 |
Intron chain level: 47.5 | 80.3 |
Transcript level: 49.2 | 73.9 |
Locus level: 86.1 | 86.7 |
Matching intron chains: 8977
Matching transcripts: 10539
Matching loci: 9015
Missed exons: 10916/54846 ( 19.9%)
Novel exons: 857/42297 ( 2.0%)
Missed introns: 8014/41091 ( 19.5%)
Novel introns: 433/28326 ( 1.5%)
Missed loci: 0/10469 ( 0.0%)
Novel loci: 334/10407 ( 3.2%)
Total union super-loci across all input datasets: 10160
14264 out of 14264 consensus transcripts written in gffcmp.annotated.gtf (0 discarded as redundant)
Help
Licence and Copyright
(c) 2018 Oxford Nanopore Technologies Ltd.
This Source Code Form is subject to the terms of the Mozilla Public License, v. 2.0. If a copy of the MPL was not distributed with this file, You can obtain one at http://mozilla.org/MPL/2.0/.
FAQs and tips
- The GFF2 files can be visualised using IGV.
- The GFF2 files can be converted to GFF3 or GTF using the gffread utility.
- The gffcompare tool can be used to compare the results of the pipeline to an existing annotation.
References and Supporting Information
See the post announcing the tool at the Oxford Nanopore Technologies community here.