FluentDNA Data Visualization Tool
This application creates visualizations of FASTA formatted DNA nucleotide data. FluentDNA generates a DeepZoomImage visualizations similar to Google Maps for FASTA files.
From 2Mbp of Genomic Sequence, FluentDNA generates this image. Changes in nucleotide bias make
individual genome elements visible even without an annotation. Add your annotation files to see how they align with the sequence features.

FluentDNA Quick Start
You can start using FluentDNA:
- Downloading and unzipping the Latest Release (Mac and Windows only).
- Open a terminal (command line) in the same folder you unzipped FluentDNA.
- Run the command
./fluentdna --fasta="FluentDNA/example_data/Human selenoproteins.fa" --runserver - Your result files will be placed in the FluentDNA directory
FluentDNA/results/. Once your private server has started, all your results are viewable at http://localhost:8000.
FluentDNA can be installed as a python module (required for Linux). See the pip install instructions for more details.
pip install --upgrade FluentDNA
Locating Results: You will need to be using the same computer the server is running on. The server will not be visible over network or internet unless your administrator opens the port.
Example Use Cases
- Simple FASTA file (DNA)
- Multi-part FASTA file (DNA)
- Annotated Genomes
- Multiple Sequence Alignment Families
- Alignment of two Genomes
- History
Simple FASTA file (DNA)
Generating a basic visualization of a FASTA file downloaded from NCBI or another source is accomplished with the following commands:
Command: ./fluentdna --fasta="FluentDNA/example_data/hg38_chr19_sample.fa" --outname="Test Simple"
This generates an image pyramid with the standard legend (insert image of legend) and nucleotide number display.
Input Data Example: FluentDNA/example_data/hg38_chr19_sample.fa
Result: Hg38 chr19 sample
It is also possible to generate an image file only that can be accessed with an image viewer using --no_webpage.
Command: ./fluentdna --fasta="FluentDNA/example_data/hg38_chr19_sample.fa" --outname="Test Simple" --no_webpage
Multi-part FASTA file (DNA)
Multi-part FASTA format includes multiple sequences in the same file. A sequence record in a FASTA format consists of a single-line description (sequence name), followed by line(s) of sequence data. The first character of the description line is a greater-than (">") symbol. Multi-part format Example:
>seq0
AATGCCA
>seq1
GCCCTAT
The following command generates a multi-part FASTA file visualization:
Input Data Example: Human Selenoproteins.fa
Command: ./fluentdna --fasta="FluentDNA/example_data/Human selenoproteins.fa"
Result: Human Selenoproteins
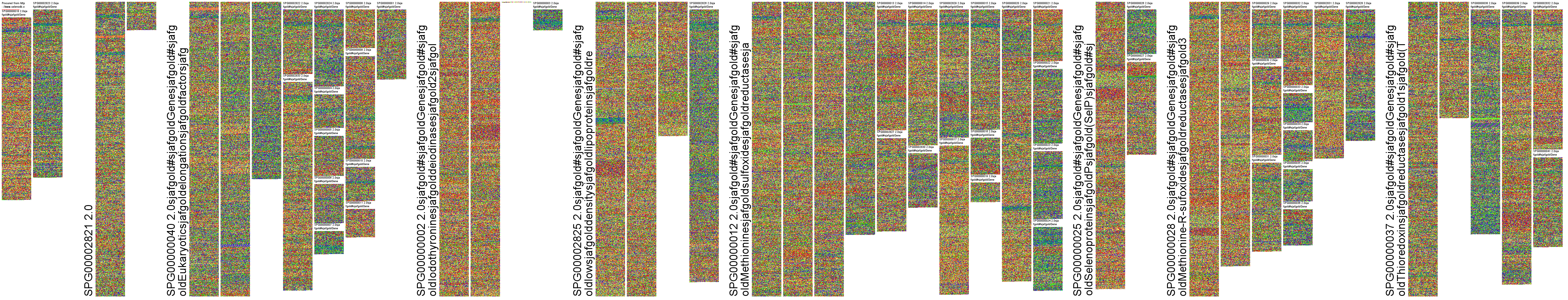
This generates a multi-scale image of the multi-part FASTA file. Note that if you don't specify --outname= it will default to the name of the FASTA file.
Using this simple command, FluentDNA can visualize an entire draft genome at once.
Result: Ash Tree Genome (Fraxinus excelsior)

Additional options - see also:
--outname--sort_contigs--no_titles--natural_colors--base_width
Annotated Genomes
By specifying --ref_annotation= you can include a gene annotation to be rendered alongside your sequence. This is currently setup to show gene introns and exons. But the features rendered and colors used can be changed in FluentDNA/Annotations.py
Command: ./fluentdna --fasta="FluentDNA/example_data/gnetum_sample.fa" --ref_annotation="FluentDNA/example_data/Gnetum_sample_genes.gff"
Input Data Example:
Result: Gnetum montanum Annotation

You can download the full Gnetum montanum files from Data Dryad.
Multiple Sequence Alignment Families
To visualize a multiple sequence alignment you need to use the --layout=alignment option to tell FluentDNA to treat each entry in a multipart fasta file as being one row of an alignment. To show many MSAs at once, just point --fasta= to a folder instead of a file.
Input Data Example:
Important Note! Make sure there are no other files in your folder besides sequence files. If FluentDNA decides on an unreasonably long "max width" it is because it picked up a non-sequence file in the folder.
Each fasta file represents one aligned protein family. The input fasta file should already be aligned with another program. Each protein is represented by one entry in the fasta file with - inserted for gap characters like this:
>GeneA_in_species1
ACTCA--ACGATC------GGGT
>GeneA_in_species2
ACTCAAAACGATCTCTCTAGGGT
Command: ./fluentdna --layout=alignment --fasta="FluentDNA/example_data\alignments" --outname="Example 7 Gene Families from Fraxinus"
This generates a multi-scale image of the multiple alignment. The multiple alignment results are sorted by gene name. For a smoother layout use --sort_contigs which will sort them by row count (copy number).
Result: Example 7 Gene Families from Fraxinus

This layout allows users to check thousands of MSAs. Here we used FluentDNA to quality check the merging software for 2,961 putative gene families: Fraxinus Homologous Gene Groups
Alignment of two Genomes
This generates an alignment visualization of two genomes (A and B). Since this is a whole genome alignment the files are very large and not included in the FluentDNA installation. Download each of these files and unzip them into a local folder called data/.
Input Data Download Links (unzip these into data folder):
- Human Genome FA File (hg38.fa)
- Alignment LiftOver Chain File (hg38ToPanTro5.over.chain)
- Chimpanzee Genome FA file (panTro5.fa)
Command: ./fluentdna --fasta=data/hg38.fa --chainfile=data/hg38ToPanTro5.over.chain --extrafastas data/panTro5.fa --chromosomes chr19 --outname="Human vs Chimpanzee"
This generates a multi-scale image of the alignment. There are 4 columns in this visualization:
- Column 1. Genome A (gapped entire DNA of genome A)
- Column 2. Genome A (unique DNA of A)
- Column 3. Genome B (unique DNA of B)
- Column 4. Genome B (gapped entire DNA of genome B)
Result: Human vs Chimpanzee_chr19 (natural colors)

Figure 1 in the paper can be seen at Nucleotide Position 548,505 which corresponds to HG38 chr19:458,731. The difference in coordinates is due to the gaps inserted for sake of alignment.
Note: Whole genome alignment visualizations can be processed in batches, one visualization per chromosome. Simply specify each of the reference chromosomes that you would like to generate. --outname will be used as a prefix for the name of the folder and the visualization. For example, the above command generates a folder called "Human vs Chimpanzee_chr19".
History
This project is a fork of the C# FluentDNA developed at Concordia University. https://github.com/photomedia/DDV/
DDV Licence: https://github.com/photomedia/DDV/blob/master/DDV-license.txt
Support Contact
If you run into any problems or would like to use FluentDNA in research, contact me at [email protected]. I'm happy to support my own software and always interested in new collaborations.